-

-

- Мои разделы
-
 Мои друзья(0)
Мои друзья(0) -
Мои фото(0)
-
Мои дневники(0)
-
Моя музыка(0)
- Разделы сайта
- Раздел сервисы и загрузок 14
- Раздел развлечения 18
- Раздел онлайн игр 76
- ✴Банды сайта❗✴ (1/3)
- ⭐Лидеры дня❗⭐ (0)
- ☀День рождения❗☀
- ☀Новости сайта❗☀ (21)
- ⭐Топ пользователей❗⭐
- ❤Даты праздников❗❤ 255/152
- ⭐Фотогалерея сайта❗⭐ (106)
- ☀Статусы обитателей ❗☀ (19)
- ❤Дневники сайта❗❤ (1)
- Микроблоги 6
- ❇Форум сайта❗❇ (55/90)
- ⭐Файловый обменник❗⭐ (3)
- ✴Анаграмма❗✴ 0 человек
- ☀Администрация сайта❗☀ (1)
- ✳Викторина❗✳ 0 человек
- ☀Чат сайта❗☀ (0 человек)
- ❤Бракосочетания❗❤ (2 )
- ❤Война полов❤
- ❤VIP Знакомства❤ 264
- ❤Обитатели сайта❗❤ М :136 | Ж :128 +1
- ☀Острослов☀
- Сказочный бонус (83)
- ✴Ежедневный подарок❗✴ (27)
- Аукцион Джинов
- Зарница (3)
- ❤Лотерея❤
- Спортлото 6 из 36
- Русская рулетка (35)
- Моя удача
- Дополнительно
-
Выход
 Получить зарплату💸💰💳
Получить зарплату💸💰💳
 Банк сайта
Банк сайта💌mirsoc.ru
Математики придумали архиватор для ДНК
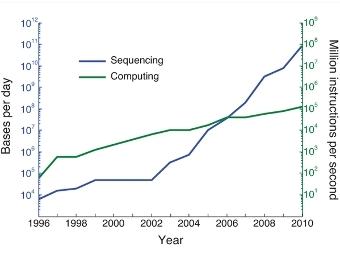
Математики из Массачусетского технологического института предложили новый способ хранения и обработки данных о последовательностях ДНК. Он должен помочь справиться с наплывом данных от все большего числа прочитанных геномов. Работа с описанием нового алгоритма опубликована в журнале Nature Biotechnology, а ее краткое содержание можно прочитать на сайте института.
Алгоритм основан на том, что последовательности ДНК между всеми организмами в той или иной степени схожи, а наибольший интерес для ученых представляют различия. Поэтому, по словам авторов, хранить и обрабатывать следует не сами последовательности, а их отличия друг от друга.
Если, например, поиск определенной последовательности в геноме некоторого организма уже проводился, то поиск той же последовательности в новом геноме следует проводить не по всей последовательности, а только в тех местах, где новый геном отличается от старого. Это позволяет существенно снизить время поиска последовательностей и нагрузку на вычислительные центры. Разница в длительности вычислений между старым и новым алгоритмом зависит от количества уже прочитанных геномов - чем их больше, чем труднее искать по-старому и тем очевиднее преимущества нового алгоритма.
Упор на поиск различий в близких геномах соответствует современному развитию биологии. С одной стороны, в последнее десятилетие резко уменьшается стоимость секвенирования. Из-за этого скорость роста данных о последовательностях ДНК уже превышает экспоненциальную. С другой стороны, по мере увеличения количества прочитанных геномов доля совершенно уникальных последовательностей уменьшается. Прочитанные геномы все больше походят друг на друга. Например, в ближайшее время биоинформатики ожидают массового наплыва данных от проектов по секвенированию ДНК тысяч отдельно взятых людей, позвоночных, насекомых.
